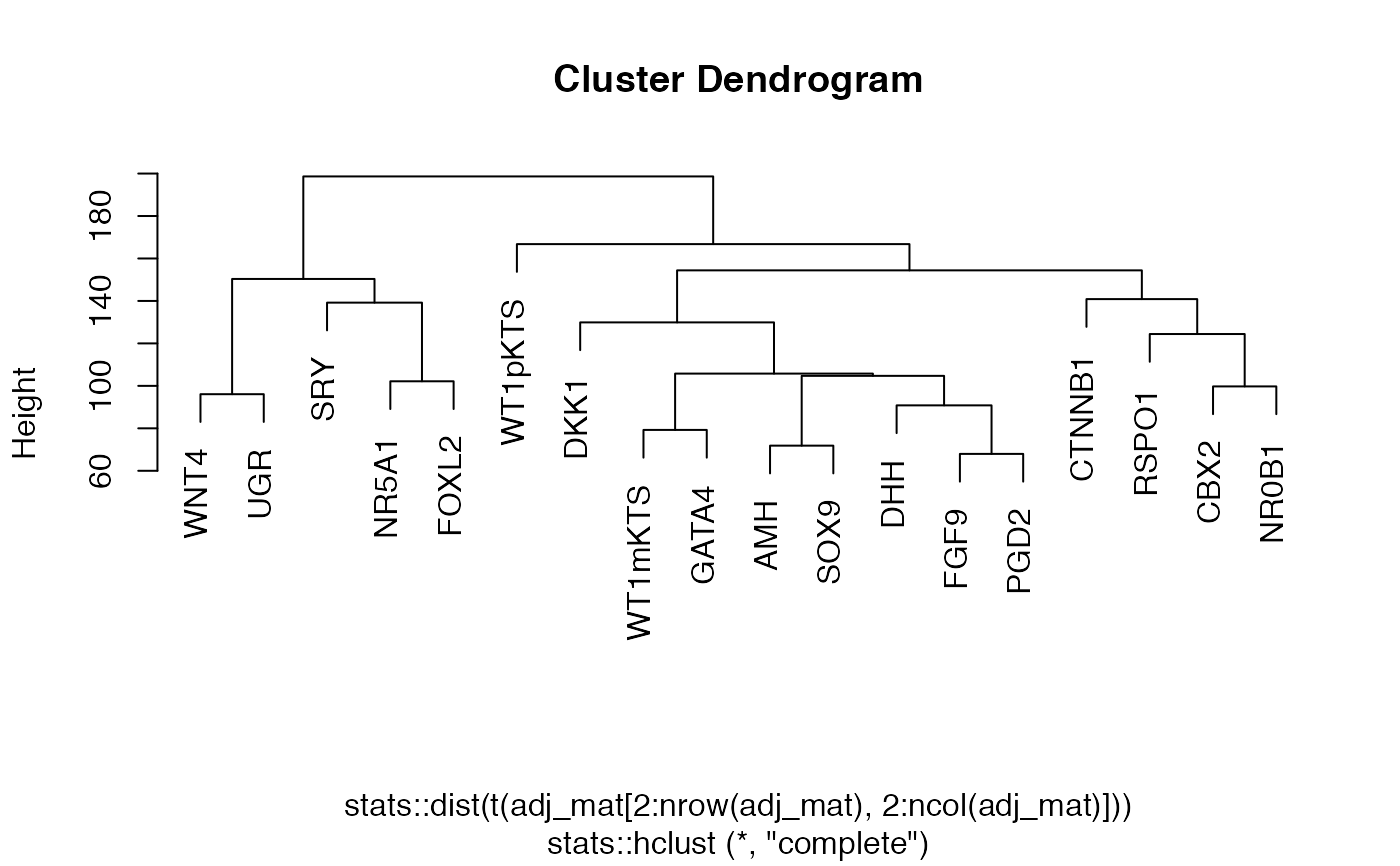
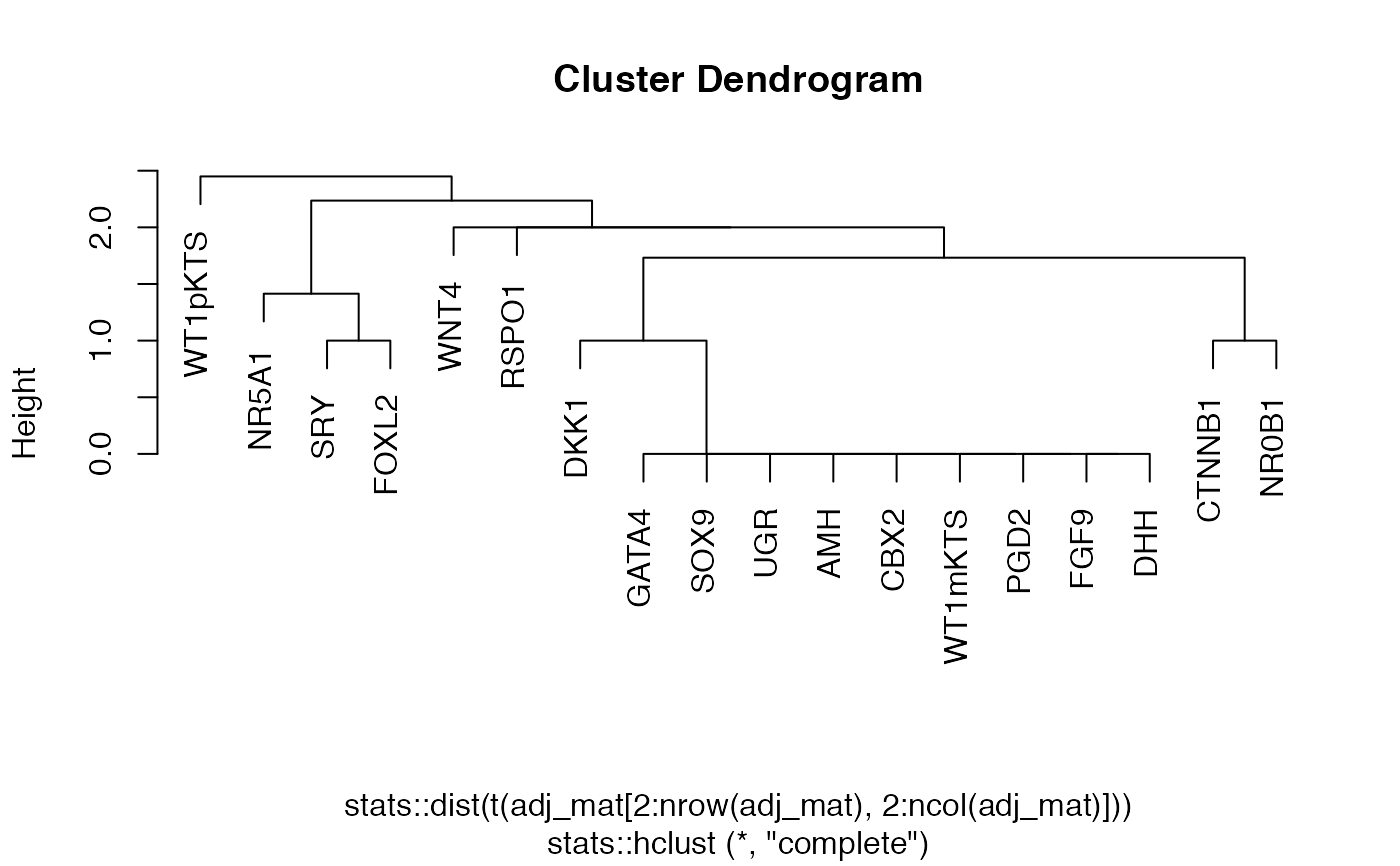
Matrix convert to binary
network_make_binary.RdThis function takes a weighted adjacency matrix and separates the components of the matrix into weights and presence/absence matrices making use of a threshold.
network_make_binary(adj_mat, threshold, print_message = TRUE)
Arguments
| adj_mat | adjacency matrix to be converted. |
|---|---|
| threshold | we recommended to use
|
| print_message | a logical value indicating whether or not a threshold message shall be printed. |
Author
Sergio Vasquez and Hajk-Georg Drost
Examples
# path to PPCOR output file ppcor_output <- system.file('beeline_examples/PPCOR/outFile.txt', package = 'edgynode') # import PPCOR specific output ppcor_parsed <- ppcor(ppcor_output) # rescale the matrix ppcor_rescaled <- network_rescale(ppcor_parsed)#># make the binary matrix ppcor_binary <- network_make_binary(ppcor_rescaled, 70)#>